- Deli
-

-

-

- Prenesi PDF
-

- Natisni
-

Predvajaj avdio zapis članka

Ko so bile leta 2024 podeljene Nobelove nagrade, je marsikdo presenečeno ugotovil, da so bile tako za fiziko kot za kemijo nagrajene raziskave povezane z umetno inteligenco. A to ni naključje – znanost postaja vse bolj interdisciplinarna, meje med klasičnimi področji pa se brišejo. Ena od disciplin, ki se je v zadnjih desetletjih zelo razvila prav zaradi računalniških metod, je računska kemija. Če laboratorijski eksperiment omogoča neposredno opazovanje kemijskih procesov, potem lahko računalniške molekulske simulacije razumemo kot virtualni mikroskop, ki omogoča vpogled v svet molekul in atomov. Veliko kemijskih reakcij in bioloških procesov poteka na časovnih in prostorskih skalah, ki so premajhne za tradicionalne eksperimentalne metode. S simulacijami pa lahko spremljamo dogajanje na ravni posameznih atomov, kar nam omogoča razumevanje mehanizmov, ki bi sicer ostali skriti.
Takšen pristop ne prinaša le boljšega razumevanja osnovnih procesov v kemiji in biologiji, temveč je ključen tudi za razvoj novih materialov, katalizatorjev in zdravil. Namesto da bi se zanašali zgolj na drage in dolgotrajne laboratorijske poskuse, lahko danes s pomočjo simulacij že vnaprej z visoko natančnostjo napovemo, katerim spojinam bi bilo smiselno posvečati eksperimentalno pozornost. To pripomore k hitrejšemu napredku in bolj ciljno usmerjenemu raziskovanju, zaradi česar je računska kemija nepogrešljivo orodje v sodobni znanosti.
Računalniška infrastruktura je ključna za izvajanje molekulskih simulacij
Molekulske simulacije so računsko zahtevne, saj pri sistemih, ki pogosto vsebujejo več tisoč atomov, vsak časovni korak zahteva izračun milijonov medsebojnih interakcij. Poleg tega je treba obdelati ogromne količine podatkov, ki nastajajo med simulacijami. Predstavljajte si, da želite opazovati, kako se zdravilo veže na virusni protein. Molekule so v nenehnem gibanju, in da bi razumeli njihovo dinamiko, moramo na vsakem časovnem koraku simulacije izračunati milijone interakcij med atomi. To pomeni, da se v eni sami simulaciji lahko ustvari terabajte (tj. bilijone bajtov) podatkov. Razvoj zmogljive računalniške infrastrukture ima zato ključno vlogo za razvoj simulacij.
Računalniške gruče, ki so skupine več računalnikov, povezanih med seboj in delujočih kot celota, so omogočile, da se naloge, ki bi bile pretežke za en sam procesor, razdeli med tisoče procesorjev, ti pa jih uporabljajo za sočasno obdelavo podatkov. Pomemben napredek so prinesle tudi grafične procesne enote (GPU), ki so bile sprva zasnovane za obdelavo 3D‑grafike, danes pa so ključna strojna oprema za molekulske simulacije in raziskave na področju umetne inteligence. GPU-procesorji omogočajo izvajanje tisoče operacij hkrati, kar lahko simulacije v primerjavi z običajnimi CPU-procesorji pospeši od 100- do 1000-krat, kar pomeni veliko hitrejše in bolj učinkovite raziskave.
Raziskovalci po vsem svetu uporabljajo za simulacije kompleksnih bioloških sistemov superračunalnike, npr. Frontier in Anton 3 v ZDA, JUWELS v Nemčiji ter Fugaku na Japonskem. Eden najbolj posebnih je superračunalnik Anton 3, ki je specializiran izključno za molekulske simulacije. Gre za naslednika prejšnjih verzij Anton 1 in Anton 2, ki jih od leta 2008 razvija podjetje D. E. Shaw Research. Zgodba o njihovem nastanku je prav navdušujoča, saj je prof. David E. Shaw sprva zaslovel v finančnem svetu, kjer je z naprednimi algoritmi napovedoval gibanje borznih trgov, nato pa je svoje znanje preusmeril v biološke raziskave. Njegova skupina je zasnovala superračunalnik Anton, ki omogoča raziskave, kakršne bi bile nekoč nepredstavljive. Z njegovo pomočjo lahko simuliramo zvijanje proteinov, napovedujemo učinkovitost novih zdravil in celo modeliramo delovanje celotnih celic.
Tudi Slovenija ima več računskih centrov. Najmočnejši superračunalnik je HPC Vega, poimenovan po slavnem matematiku Juriju Vegi. Na Kemijskem inštitutu v Ljubljani deluje nekoliko manjši Ažmanov računski center, ki se večinoma uporablja za izvajanje molekulskih simulacij (Slika 1). Poimenovan je po prof. dr. Andreju Ažmanu (1937–1980), enem izmed pionirjev računske kemije v Sloveniji. V letu 2025 je Slovenija pridobila sredstva za postavitev novega superračunalnika, ki bo omogočil še hitrejše izvajanje simulacij in ki naj bi začel delovati leta 2027.

Slika 1: Molekulske simulacije zahtevajo zmogljivo računalniško infrastrukturo. Eden takšnih sistemov je Ažmanov računski center na Kemijskem inštitutu v Ljubljani.
Od eksperimenta do algoritmov – iskanje tridimenzionalne strukture molekul
Če želimo molekulo preučevati s pomočjo računalniških metod, moramo najprej vedeti, kako je videti – torej poznati njeno tridimenzionalno strukturo. Ta določa, kako se bo molekula vedla v različnih pogojih in kakšne bodo njene interakcije z drugimi molekulami. V kemiji govorimo o konformaciji, ki opisuje natančen položaj vseh atomov v molekuli (Slika 2).
Tridimenzionalno strukturo molekule lahko določimo eksperimentalno, in sicer s tehnikami, ki omogočajo pridobitev nekakšnega »posnetka« njene 3D-oblike oziroma konformacije. Rentgenska kristalografija in jedrska magnetna resonanca (angl. Nuclear Magnetic Resonance oz. NMR) sta dolgo časa veljali za zlati standard pri določanju molekularnih struktur. V zadnjem desetletju pa se je pojavila nova metoda – krioelektronska mikroskopija (angl. Cryogenic Electron Microscopy oz. cryo-EM), ki uporablja elektrone in omogoča izjemno natančne slike biomolekul, kot so proteini in molekula DNK (Slika 2).
Včasih so molekule prevelike, preveč kompleksne ali preveč nestabilne, da bi lahko eksperimentalno določili njihovo 3D-strukturo. Tu na pomoč priskočijo računalniške metode, ki omogočajo napovedovanje tridimenzionalne strukture (Slika 2). Največji korak naprej na tem področju predstavlja program AlphaFold, ki za zelo natančne napovedi strukture proteinov uporablja umetno inteligenco. Razvili so jo pri podjetju DeepMind (del Googla) in velja za enega največjih prebojev v biologiji v zadnjih desetletjih. Za razvoj programa AlphaFold sta raziskovalca Demis Hassabis in John Jumper leta 2024 prejela tudi Nobelovo nagrado za kemijo.
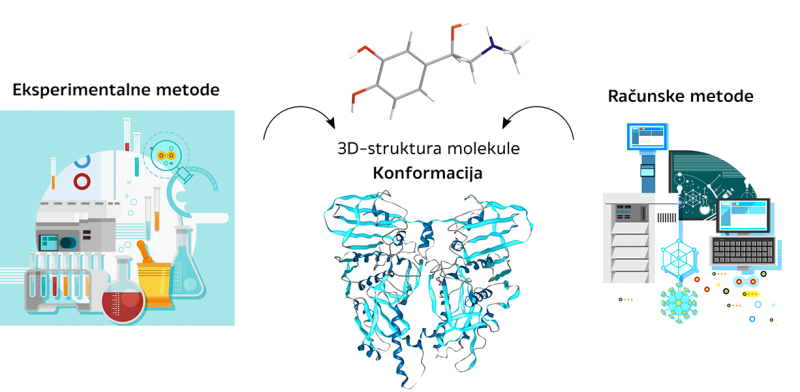
Slika 2: Prikaz eksperimentalnih in računskih metod za določitev tridimenzionalne strukture molekul, kot so proteinska kristalografija, NMR-spektroskopija, krioelektronska mikroskopija in AlphaFold. Deli slike so vzeti s spletnega mesta Macrovector/Freepik.
Različne poti do energije molekule – od kvantne mehanike do grobozrnatih modelov
Ko z računalniškimi simulacijami modeliramo molekule, je eden prvih parametrov, ki jih izračunamo, energija molekule, saj je ta neposredno povezana s stabilnostjo določene molekulske konformacije. Nižja kot je energija, bolj ugodna in stabilna je posamezna konformacija. Vendar pa so izračuni pri večjih molekulah zahtevnejši, zanje pa uporabljamo različne pristope, ki se med seboj razlikujejo po natančnosti, hitrosti in računski zapletenosti.
Najnatančnejši način temelji na kvantni mehaniki, ki atome in njihove sestavne delce (protone in nevtrone v jedru ter elektrone, ki jih obdajajo) obravnava kot kvantne delce (Slika 3). Namesto da bi molekulo preprosto videli kot skupek krogel (atomov) in vzmeti (kemijskih vezi), kot si jo pogosto poenostavljeno predstavljamo, jo v tem pristopu opisujemo z matematičnimi (valovnimi) funkcijami, ki določajo verjetnost položaja kvantnih delcev. Ta pristop je zelo natančen, vendar tudi zelo računsko zahteven, zato je kvantna mehanika primerna za raziskovanje manjših molekul ali posameznih kemijskih reakcij.
Za večje molekule, kot so proteini in DNK postane kvantna mehanika prepočasna, zato uporabimo molekulsko mehaniko. Tu molekulo predstavimo kot sistem kroglic (atomov), povezanih z vzmetmi (kemijskimi vezmi), pri čemer vsak atom obravnavamo kot celoto brez upoštevanja njegove sestave. Ta metoda temelji na klasični fiziki in omogoča simulacije veliko večjih sistemov, čeprav je zaradi tega pri opisovanju podrobnih medatomskih interakcij malo manj natančna (Slika 3).
Pri izjemno velikih sistemih, kot so proteinski kompleksi, virusni delci ali polimeri, pa tudi molekulska mehanika postane prepočasna za izračun energije. Tu nastopijo grobozrnati modeli, ki več atomov združijo v eno večjo enoto (Slika 3). Namesto da bi sledil vsakemu posameznemu atomu, ta model spremlja obnašanje celotnih sklopov. Tako lahko simuliramo izredno velike biološke sisteme in celo nanodelce, vendar le na bolj splošni ravni.
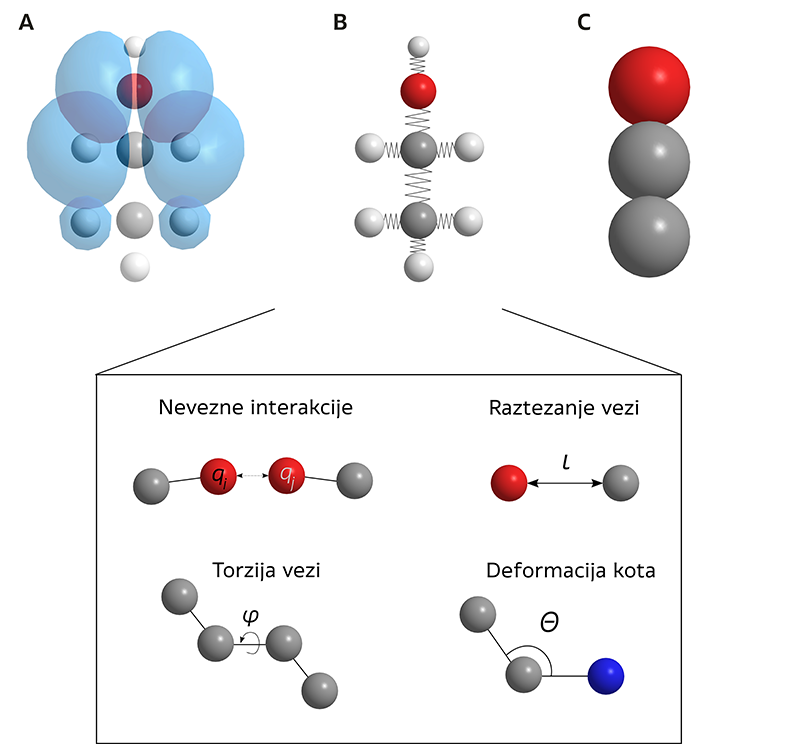
Slika 3: Prikaz različnih modelov (kvantna mehanika, molekulska mehanika, grobozrnati modeli) za računanje energije molekul na primeru molekule etanola
Izbira prave metode za izračun energije je pri računalniških simulacijah ključnega pomena, saj mora raziskovalec najti ustrezno ravnovesje med natančnostjo, hitrostjo in velikostjo sistema. Za nekatere raziskave je potrebna izjemna natančnost, na primer pri preučevanju kemijskih reakcij ali interakcij med zdravilnimi učinkovinami in njihovimi tarčami. V drugih primerih je pomembnejši hiter pregled širših molekularnih procesov. Raziskovalci pogosto kombinirajo različne pristope, da dosežejo najboljše rezultate za specifične naloge.
Molekulske simulacije – računalnik kot virtualni mikroskop za pogled v molekulski svet
Vsaka molekula ima svojo energijsko površino, ki prikazuje, katera od njenih oblik oziroma konformacij je najstabilnejša (Slika 4). Vendar pa se število takih oblik hitro povečuje, še zlasti pri večjih molekulah. Za ilustracijo si zamislimo primer, kjer želimo raziskati vse možne konformacije peptida, ki vsebuje deset aminokislin. Kot poenostavitev predpostavimo, da ima kar vsaka aminokislina glede na število različnih rotacijskih kotov okoli vezi 15 rotacijskih parametrov. V tem primeru bi morali preiskati 1510 konformacij. Če predpostavimo, da za izračun energije ene konformacije potrebujemo 0,1 sekunde, potem bi skupni čas za preučitev vseh možnih konformacij znašal 667.411 dni oziroma kar 1828 let. Zato se izračuna sistematično ne moremo lotiti niti z najzmogljivejšimi superračunalniki. Da bi to časovno obdobje skrajšali, računalniki uporabljajo posebne tehnike, ki omejijo število preiskanih oblik na tiste, ki so najbolj verjetne in pomembne.
Za iskanje najstabilnejših konformacij molekul uporabljamo energijsko minimizacijo, pri kateri računalnik poišče obliko molekule z najnižjo možno energijo (Slika 4). Kadar želimo poleg minimalne strukture molekule raziskati več stabilnih oblik, ki so prav tako pomembne, je zelo uporabna metoda Monte Carlo. Ta temelji na naključnem spreminjanju položajev atomov, da se tako preveri, ali bi to pripeljalo do nove stabilne strukture (Slika 4). Vendar pri tej metodi ne spremljamo gibanja molekule skozi čas, temveč najbolj stabilne oblike iščemo z naključnimi spremembami.
Za spremljanje gibanja molekule skozi čas uporabljamo metodo molekulske dinamike. Računalnik s pomočjo Newtonovih zakonov gibanja (spomnimo se znane enačbe F = m · a, kjer je (v primeru molekul) F sila na posamezen atom, m masa atoma in a pospešek) premika atome molekule v zelo kratkih časovnih korakih. Za te izjemno hitre premike uporabljamo časovno enoto femtosekunda (ena milijoninka milijoninke sekunde). Simulacija molekulske dinamike nam tako omogoča, da lahko opazujemo premikanje in spreminjanje atomov molekule skozi čas, kar je sicer nemogoče spremljati s prostim očesom (Slika 4).
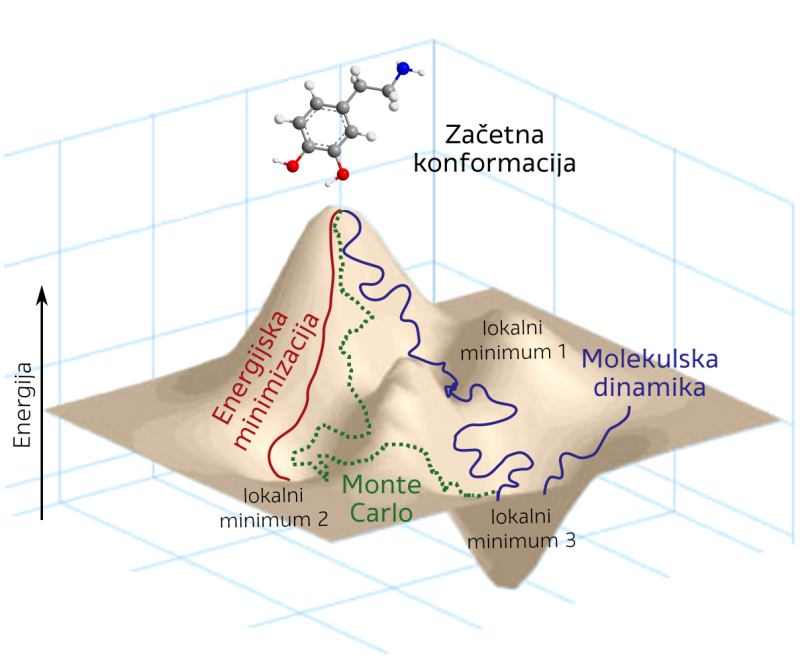
Slika 4: Poenostavljen prikaz energijske površine molekule dopamina in različnih pristopov za raziskovanje njenih struktur oz. konformacij. 1) Energijska minimizacija poišče najstabilnejšo konformacijo molekule. 2) Metoda Monte Carlo uporablja naključne spremembe za iskanje različnih stabilnih oblik molekule. 3) Simulacija molekulske dinamike omogoča sledenje gibanju molekule skozi čas.
Prihodnost virtualnih laboratorijev – razvoj novih metod in sinergija z eksperimentom
Molekulske simulacije so v zadnjih desetletjih doživele izjemen napredek, a njihov polni potencial še vedno ni povsem izkoriščen. V prihodnosti lahko pričakujemo razvoj še naprednejših in učinkovitejših metod, ki bodo omogočile raziskovanje večjih in kompleksnejših bioloških sistemov, kot so celice, tkiva ali celo organi. Z napredkom v simulacijah bomo tudi hitreje razvijali zdravila za kompleksne bolezni, kot je na primer rak, ali pa ustvarjali nove, bolj trajnostne materiale, ki bodo zmanjšali okoljski odtis. Molekulske simulacije bodo postale še učinkovitejše orodje za usmerjanje raziskav, saj bodo omogočale bolj premišljeno načrtovanje eksperimentov, zmanjšanje stroškov in hitrejše odkrivanje molekul z želenimi lastnostmi, kar bo pripomoglo k hitrejšemu napredku v znanosti in tehnologiji.
Ena od najbolj obetavnih novosti bo uporaba umetne inteligence, ki bo znatno izboljšala hitrost in natančnost simulacij. Z naprednimi algoritmi, kot so nevronske mreže, bo mogoče bolje napovedovati, kako molekule medsebojno delujejo, hkrati pa zmanjšati za to potrebne računalniške kapacitete. To pomeni, da bodo raziskave lahko obravnavale večje sisteme z večjo natančnostjo in v krajšem času.
Kljub hitremu razvoju simulacij pa te ne bodo popolnoma nadomestile eksperimentalnega dela v laboratorijih. Novi modeli bodo omogočali hitrejše in natančnejše napovedi, vendar bodo eksperimentalne potrditve še vedno ključne za preverjanje računskih ugotovitev: opraviti bo treba biokemijske teste, preveriti stabilnost spojin v realnih pogojih ter ovrednotiti njihovo dejansko delovanje v bioloških sistemih. Prihodnost računske kemije nakazuje še močnejšo sinergijo z eksperimentalnim delom, kjer bo vsaka metoda prispevala edinstven vpogled v svet molekul in pripomogla k boljšemu razumevanju naravnih procesov.